
Manapság komolyabb publikációk nem elégszenek meg egyféle vizsgálati típussal, az összetett biológiai rendszerek feltérképezéséhez több szekvenálási módszer eredményét ötvözik. Ezek közül talán a legegyszerűbb az eQTL, ahol a mutációs és expressziós adatokat vetik össze.
A módszer lényege, hogy egy genomi pozíció homozigóta recesszív, heterozigóta, homozigóta domináns genotípusa egy gén expresszióját befolyásolja. Ha tehát a három genotípushoz tartozó expressziós értékekre egy lineáris modellt alkalmazunk, statisztikailag megállapíthatjuk a kapcsolat erősségét. Az elmondottakat sokkal jobban szemlélteti a következő ábra:
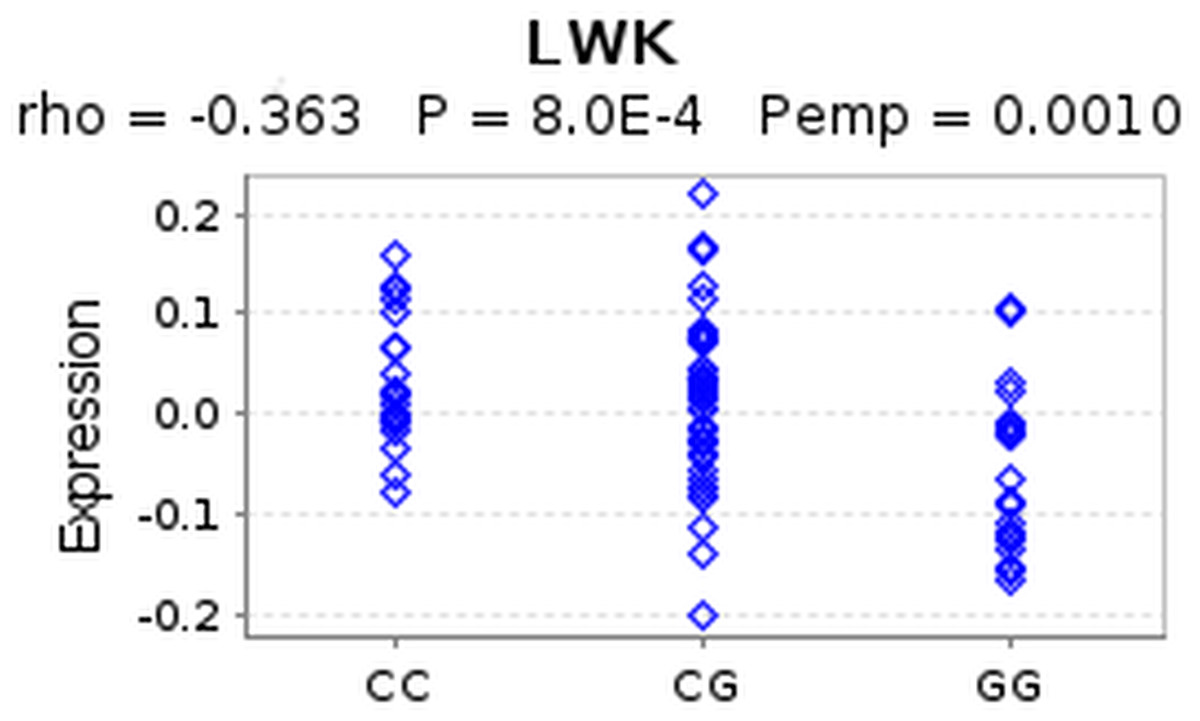
A CC genotípus mintáiban az expressziók magasabbak, mint a GG genotípus esetén.
Az eQTL számítása igen idő igényes. Minden egyes SNP és gén expresszió kombinációra ki kell számolni a korábban említett lineáris modellt. Természetesen a kellő statisztikai erő eléréséhez rengeteg mintára van szükség. A régebbi programok nem ritkán hónapokig futottak.
Attól függően, hogy milyen az SNP és az expresszálódó gén távolsága, megkülönböztetünk cisz, illetve transz eQTL-t. Előbbi esetben a két jellemző ugyan azon a kromoszómán fordul elő, utóbbinál különbözőn.
A fejlettebb programok képesek kovariánsokat is figyelembe venni. Képzeljük el azt az esetet, amikor az egyik genotípus véletlenül csak azonos típusú mintákból származik. Például valami miatt csak nőkben található meg. Ebben az esetben nem az SNP hatását vizsgáljuk, hanem valami más jelenségét. A kovariánsok felhasználásával ezek a nem kívánt események kiszűrhetőek.
Én eddig csak a MatrixEQTL csomagot használtam R-ben. A program minimum öt mátrixot vár bemenetként. Az első az SNP mátrix. A genotípust a 0, 1, 2 számokkal jelölhetjük. A mintákat az oszlopokban kell feltüntetni, míg a sorok az egyes SNP-k, egyedi azonosítóval. Hasonlóan épül fel az expressziós mátrix is, de itt a normalizált expressziót adjuk meg a cellákban. Mindkét fájlnak van egy genomi lokalizációt tartalmazó párja. A kettő között a különbség, hogy az SNP egy pozíciót tartalmaz, míg a géneknél régiót adunk meg. Ez alapján tudja a program meghatározni, hogy cisz vagy transz helyzetű-e a kapcsolat.
Végezetül kell egy kovariáns táblázat is. A sorok a kovariánsok, míg az oszlopok a minták. Minden mátrixban a mintáknak ugyan abban a sorrendben kell lennie és a pozíció fájlokban is olyan sorrendben kell feltüntetni az SNP-ket/géneket, ahogy a másik mátrixban is megtalálhatóak.
A programot könnyű használni, gyorsan lefut, folyamatosan fejlesztik. Nem véletlen, hogy a GTEx is ezt használja.