
A döntési fák alapötletét egy viccel lehet szemléltetni:
Az öreg székely a vacsora asztalnál ül a feleségével és a fiával. Egyszer csak egy hatalmas szellentés hallatszik. Az öreg megkérdezi:
- Asszony, te voltál?
- Nem.
- Fiam, te voltál?
- Nem.
- Akkor én - vonja meg a vállát az öreg.
Vagyis a döntési fák egy sor bináris feltétel összessége. Első sorban osztályozásra használják, ahol a feltételek (és ezáltal a döntések) végén egy kategória található. A való életben, anélkül hogy tudatában lennénk, számtalan helyen alkalmazzuk: ehető gombák meghatározásánál, annak eldöntésére, hogy át fogunk-e menni a vizsgán (ha ez a tanár vizsgáztat, bármennyit is tudok, nem fogok átmenni), fog-e esni az eső (párás a levegő, sötét felhők vannak az égen, stb). De a barchópa nevű játék lényege is a helyes döntési fa felállítása.
A fenti példákból is látszik, egy döntés csak bizonyos eséllyel jósolja meg a választ. Minél több döntésünk van, annál pontosabb lesz a válasz. Természetesen ha a rendelkezésre álló adatok nem elég pontosak, nem terjednek ki mindenre, akkor a döntések számának növelése nem fog pontosabb választ adni. Tipikusan ez a helyzet, ha bi- vagy multimodális eloszlású adat alapján akarunk osztályozni.
Hogy mindez érthetőbb legyen, vegyük azt a példát, amikor a testmagasság alapján akarjuk eldönteni egy emberről, hogy férfi vagy nő. A férfiak általában magasabbak, de egy női magasugró besorolása már nehezebb.
Épp ezért léteznek különböző mérőszámok, amelyek megmutatják, hogy az adott döntési ág mennyire pontos. Ilyen például a Gini tisztasági szám (Gini impurity). Azt adja meg, hogy az adott döntési ágat figyelembe véve egy véletlenszerűen kiválasztott elem, milyen eséllyel kerül rossz kategóriába. Minél kisebb, annál jobb.
Nézzünk egy példát! Tegyük fel, a tüdő adenokarcinómát és sejtes karcinómát akarunk elkülöníteni génexpresszió alapján. Meg akarjuk tudni, mennyi gén alapján lehet eldönteni, hogy milyen típusú betegségben szenved valaki. A döntési fa elkészítéséhez a scikit-learn Python csomagot fogjuk használni.
Először is töltsük le a TCGA-LUAD és TCGA-LUSC adatokat a innen. Csak a már normalizált expresszióra lesz szükségünk. Ezek az adatok regisztráció nélkül is elérhetőek. Készítsünk két könyvtárat az egyes rák típusoknak és mentsük a fájlokat oda. Példaként itt az egyik képernyőkép, amit látni kell, ha mindent jól állítottunk be.
Ezután készítünk egy mátrixot, ahol a sorok a páciensek lesznek, az oszlopok a gének. Kis túlzással minden fájl egy páciensnek felel meg (valójában egy páciensből több adat is lehet, de most ezzel nem foglalkozunk), a fájlokban a gének ugyan olyan sorrendben vannak, ami megkönnyíti a feldolgozást.
def getMatrix(root):
files = glob.glob(root + "/*/*.gz")
matrix = list()
for i in files:
expfile = gzip.open(i)
matrix.append([])
for lines in expfile:
expression = lines.decode("utf-8").rstrip().split()[1]
matrix[-1].append(float(expression))
expfile.close()
return matrix
Ha a letöltést a gdc-client programmal végezzük, akkor az mindegyik páciensnek létrehoz egy alkönyvtárat, azért szerepel /*/*.gz a glob paramétereként. Hozzuk létre az adat mátrixot.
luadmatrix = getMatrix("luad")
luscmatrix = getMatrix("lusc")
data = luadmatrix + luscmatrix
Szükségünk lesz még egy listára, ami a jelöléseket tartalmazza, hogy a mátrix adott sora melyik betegségtípusba tartozik.
category = ['luad'] * len(luadmatrix) + ['lusc'] * len(luscmatrix)
Szükségünk van még a gének neveire. Jelen esetben ez csak EnsEMBL azonosítókra korlátozódik, de legalább nem fenyeget az a veszély, hogy kedvenc génünket nézegetjük. Mivel mindegyik fájlban ugyan olyan sorrendben vannak a gén azonosítók, ezért választunk egy tetszőleges fájlt és kiszedjük az azonosítókat belőle.
genenames = list()
expfile = gzip.open("lusc/5dd30ba1-2051-42f6-8fb4-3c49c93b8bf1/6976923e-a954-4ba0-9d1d-f97d7373ba48.FPKM.txt.gz")
for lines in expfile:
gname = lines.decode("utf-8").split()[0]
genenames.append(gname)
expfile.close()
Nagyszerű! Nincs más hátra, mint betanítani a döntési fánkat. Egy igazi adat elemző munkánál persze ellenőrizzük az adatokat, átrendezzük őket, tisztítjuk. Ha prediktív modellt akarunk építeni (jósolni akarunk valamit), akkor az adatokat szétválogatjuk tanuló és teszt adatszettre, paramétereket állítgatunk, stb. De ennek a posztnak most nem az a lényege, csak egy gyors bemutatás, ezért a teljes adatszettet felhasználjuk a tanításra.
from sklearn import tree
dectree = tree.DecisionTreeClassifier(max_depth = 5)
dectree = dectree.fit(data, category)
Viszonylag gyors, nem? Nézzük meg, mit kaptunk.
import graphviz
dot = tree.export_graphviz(dectree, out_file=None, feature_names=genenames, class_names=['luad', 'lusc'], rounded=True, filled=True)
graph = graphviz.Source(dot, format='png')
graph.render("output1")
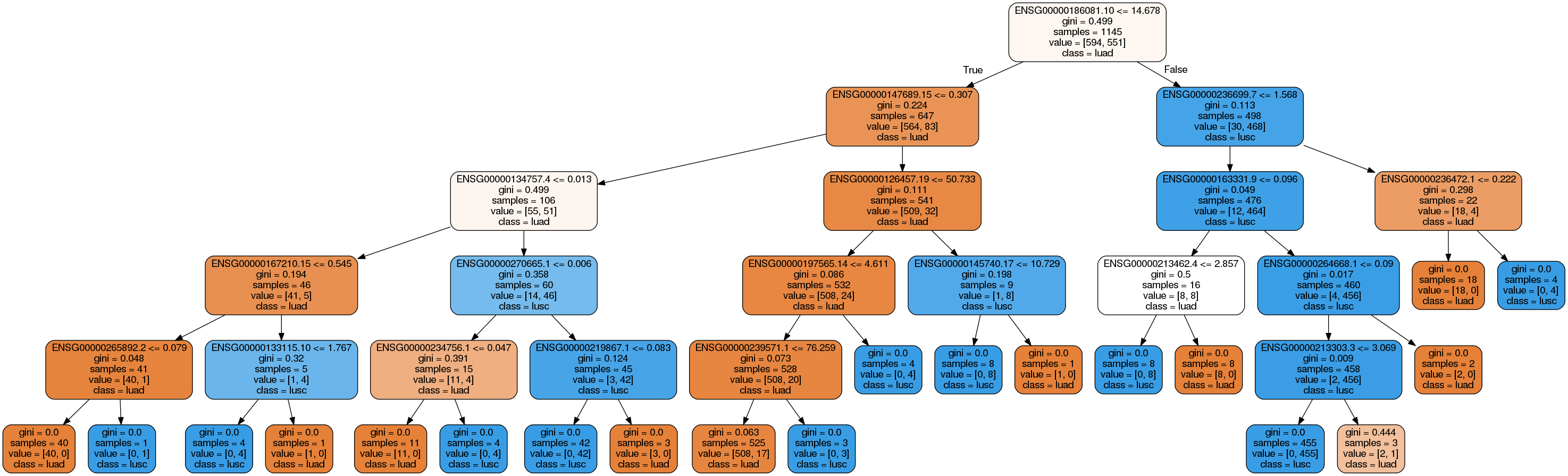
Ez már valami. Nézzük meg a fát kicsit közelebbről. Példának okáért vegyük a ENSG00000186081.10 és ENSG00000236699.7 géneket. A döntési fa szerint ha az első gén expressziója nagyobb, mint 14.678 és a másodiké kisebb vagy egyenlő, mint 1.568, akkor 30 LUAD és 468 LUSC mintát kapunk, és az algoritmusunk a LUSC kategóriába sorolja ezeket. Tehát ezen a szinten 30 minta rosszul van kategorizálva, amit a Gini tisztasáigi szám is megerősít (0.113). Amint lejjebb megyünk a fán, egyre jobb értékeket kapunk, de a mintaelem szám is csökken. Néha még 4 mintát is tovább akar bontani az algoritmus, ami remek példája a túltanításnak.
A döntési fák ugyanis hajlamosak a túltanulásra, ami azt jelenti, hogy ezen az adatszetten nagyon szép eredményeket kaptunk, de ha ráeresztenénk egy független adathalmazra, borzalmas hatékonysággal osztályozna.
A másik dolog, amit érdemes észben tartani, hogy ha az adatok transzformáción esnek át, az egész döntési fa használhatatlan lesz. Nagyon érzékeny az adatokra, amit a következő demonstrációval érzékeltetek. Távolítsuk el azokat a mintákat, ahol az átlagos expresszió 5 alatt van. Ilyen szűrést előszeretettel csinálnak expressziós adatok elemzésénél.
subsetdata = list()
subsetcateg = list()
for i in range(len(data)):
average = sum(data[i]) / float(len(data[i]))
if average > 5.0:
subsetdata.append(data[i])
subsetcateg.append(category[i])
dectree2 = tree.DecisionTreeClassifier(max_depth = 5)
dectree2 = dectree.fit(subsetdata, subsetcateg)
dot = tree.export_graphviz(dectree2, out_file=None, feature_names=genenames, class_names=['luad', 'lusc'], rounded=True, filled=True)
graph = graphviz.Source(dot, format='png')
graph.render("output2")
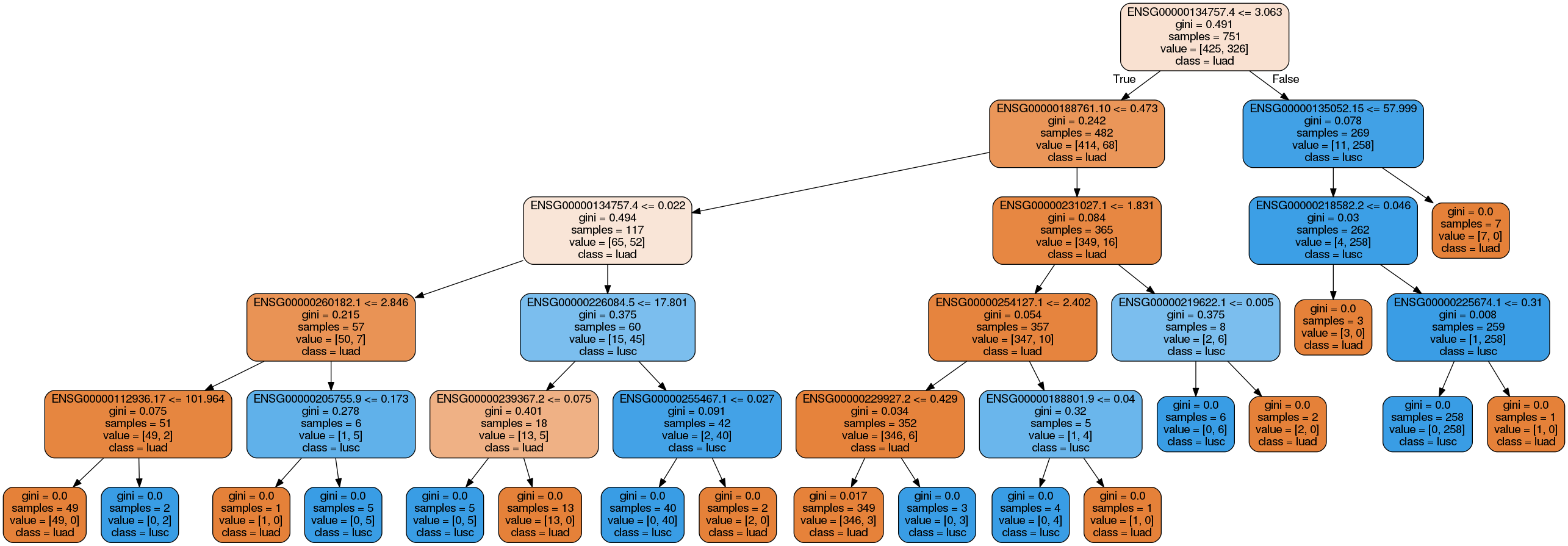
A fa teljesen más! Még a legfelső szinten is. Éppen ezért önmagában egy döntési fát ritkán használnak bármire is. Több fa viszont képes arra, amire egy nem. Ezért egy későbbi posztban a random erdőkkel fogunk megismerkedni. A Jupyter notebook itt található.
Felhasznált források: