
Elérkeztünk a burn-in lépéshez. Ennek során egy ismert szekvenciájú lambda fágot kellett megszekvenálni. Ez még a teszt része és a protokolok begyakorlására is kiválóan alkalmas. Mivel nálunk két készülék is van, elég adatot tudtam gyűjteni az elemzésekhez és alaposan begyakorolhattam, mire is kell klikkelni. Sőt, a pipettázásból is kivettem a részemet.
A könyvtár készítés egyértelmű a leírások alapján. A teljes protokollt felesleges itt részletezni, mert úgyis fantázianeveket tartalmaz (pl. motor fehérje), valamint folyamatosan változik. Volt is ebből félreértés, mert kinyomtatták a protokollt, a neten pedig időközben frissítettek. De még olyan is előfordult, hogy a kereszt hivatkozások eltérő módon frissültek, és megnehezítették a szöveg értelmezését. Ellenben felvettem mindent, a videó megtekinthető itt. Ez nem az a marketing szagú anyag, ahol előre beállított pipettákkal egy steril (vagyis használaton kívüli) laborban egy jól fésült ember végzi a lépéseket. Ez kérem a valóság: Kupleráj, szervezetlenség és hibák:
A második rész:
A minta felvétele után 6 órán keresztül ment a kis gép. A belső fórumon panaszkodtak, hogy a szekvenáló képes túlmelegedni, ezért a légkondícionálót is bekapcsoltam, amitől a hőmérséklet stabilan 37 fok volt. Mivel számítógép vásárlási igényeinket rendre visszadobják, ismét saját hardverrel támogattam a magyar kutatást. A szekvenálást is megtekintheti mindenki:
Ezzel el is érkeztünk a szekvenciákhoz. A fast5 fájlok feldolgozásához immár Loman csoportja eszközt is ad poretools néven (és sikerült megakadályozniuk, hogy az általam fejlesztett Fast5 Studio legyen az első). A cucc jó, a samtools hagyományait követi, de a gyors fejlesztésnek hála annyi hülye függősége van, hogy Ubuntun kívül nem bírtam másra feltelepiteni. (Pontosabban az első olvasás után elment a kedvem a próbálkozástól) A fast5 fájlokban 1 és 3 közötti szekvencia található a leolvasás minőségétől függően. A 2D leolvasás során ugyanis a két szálú DNS-t széttekeri és külön leolvassa az egyik szálat (template), majd a másikat (complement), végül generál egy konszenzust is (twodirections). Különböző, a szekvenálások során fellépő hibáktól függően egyik-másik szekvencia hiányozhat.
A readek hossza Poisson-eloszlást követ, nálunk a várható érték mindkét szekvenálás esetén 3000 nukleotid volt, a leghosszabb pedig elérte a 78 ezer hosszúságot. A readek száma függ az úgynevezett pólus számtól. Ez gyakorlatilag azt mondja meg, hány szekvenálás történhet párhuzamosan. A flowcell elméletileg 512 pólussal érkezik, a gyakorlatban az első szekvenálás esetben 267, a másodikban 400 pólus volt aktív. Mondanom sem kell, a négyszáz pólusos sikerült jobban.
Elméletileg a flowcell egy lemosást követően újra használható, de a nálunk járt példányok esetén a pólus szám harmadolódott és ezzel a hatákonyság is. Több módszert is kipróbáltunk, de a pólusok működés képtelenné váltak. (Időközben a fórumon egyesek a pólusok megmentésére tettek próbálkozásokat).
A readek nagyon sok hibát tartalmaznak. Átlagosan minden 3-4 nukleotid hibás, és vannak indelek is. Ami viszont érdekes, hogy a szekvencia hosszával a hibák száma nem korrelál. Kapunk itt is megbízhatósági pontszámot (quality score) minden egyes nukleotidra, de a gyakorlat azt mutatja, hogy nem jelent semmit. A hibák előfordulási aránya nem mutat összefüggést ezzel a pontszámmal. Egy illesztést itt láthatunk:
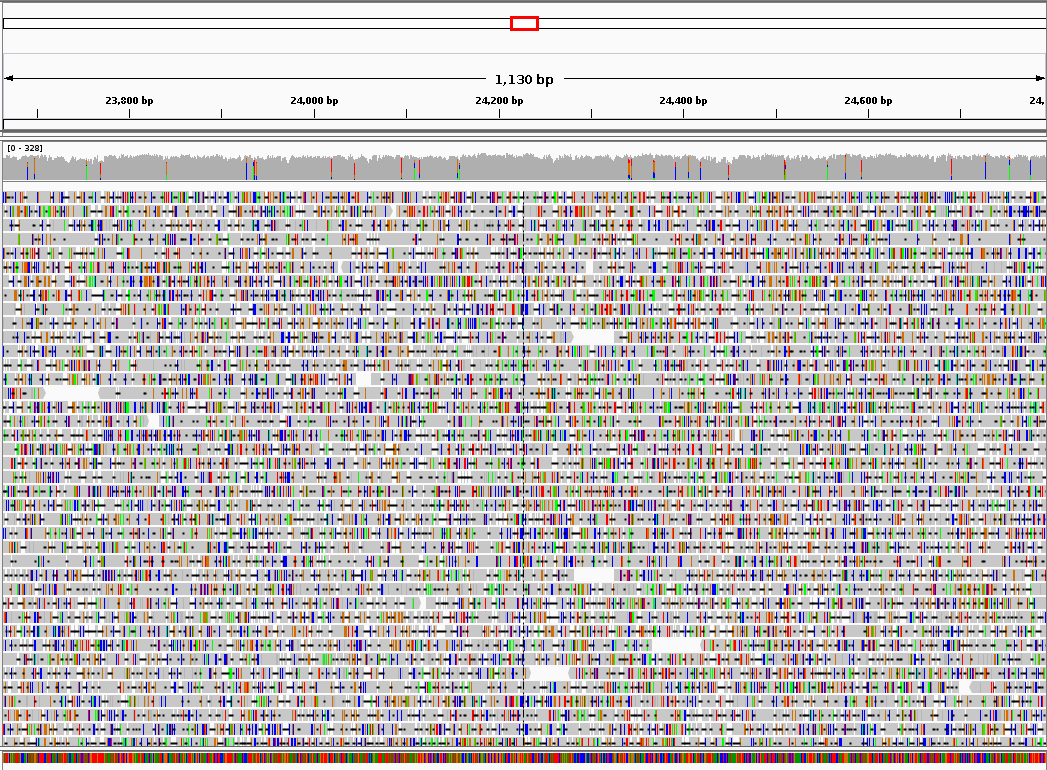
Illesztésre a Last-ot szoktuk használni, erősen megengedő módban. A hosszú szekvenciáknak köszönhetően ez mégsem probléma. Fórumon többen is összehasonlították az elérhető illesztőprogramokat, készítettek egyedi mátrixot is, hogy növelhessék a hatékonyságot, de eddig a Last tartja magát. (Azóta Ivan Sovic fejlesztett egy saját gráf alapú illesztő programot, de még nem teszteltem)
A hibákat elemezve találunk homopolimer hibát, ami nem meglepő, tekintve, hogy a DNS áthúzása a póluson valószínűleg nem konstans és a pólus konformáció változása is nehezen érzékelhető azonos nukleotidok esetén.
De-novo összeszerelés eddig kivitelezhetetlen. Illuminával történő hibrid assembly viszont ígéretesnek tűnik a SPAdes program használatával, ha a minion readeket PacBio-ként adjuk be. Erről már cikket is adtak ki.