
A nagyobb genomok összeszerelésénél sajnos a hosszú readek még nem állnak olyan szinten, hogy rutinszerűen alkalmazzuk őket. Lehet még vegyes hosszúságú inszert mérettel hibrid összeszerelést csinálni, hosszú és rövid readeket kombinálni. De jelenleg bármelyik módszert is alkalmazzuk, a homológ kromoszóma szakaszok kifognak rajtuk. Egyszerűen túl nagy a távolság hasonló genomi szakaszok között, hogy bármivel is átérjünk őket, vagy a sorrendjüket megtudjuk.
Ezért a 10X Genomics egy bárkódolási eljárással megjelöli a nagyobb fragmenteket. Szekvenálásnál ezért tudjuk, mely readek tartoznak össze. Egy plusz hozadéka is van a jelölésnek: ismerjük az anyai és apai kromoszómákat is!
Természetesen a módszer mit sem ér, ha nincs hozzá szoftver. Szerencsére a cég ezt is elkészítette, és Supernova néven teszi elérhetővé. (Természetesen miután regisztráltunk, elfogadjuk a sok spamet, stb.)
A Supernova egy, sok processzoros rendszeren fut, minimum 32 maggal és 256 vagy 512 MB RAM memóriát használnak (genom mérettől, repetitív szakaszok hosszától függően), klaszterek szóba sem jöhetnek. Saját feladat ütemezővel rendelkezik, amit a cég fejleszt Go nyelven. A neve Martian.
A Supernova másik érdekessége, hogy képes közvetlenül az Illuminás BCL fájlokon futni, ezért függőségként kell neki a bcl2fastq.
A program paraméterezése elég egyszerű, igazából a legtöbb magához a bcl2fastq-hoz kapcsolódik. Futás közben rengeteg fájlt és könyvtárat dobál szét, teljesen átláthatatlan, hol is tart. A logok semmit mondóak. A folyamat nyomon követésére a projekt helyén található ASSEMBLER_CS/_ASSEMBLER könyvtárban található alkönyvtárak adják a legjobb támpontot, mert ezek a nevek megegyeznek a dokumentációban található lépésekkel.
A végén rengeteg bináris fájl lesz az eredmény, amiből egy újabb lépéssel lesz FASTA fájl. Négy lehetséges kimenet van. Az első, amikor minden darabkát visszaad. A második a kisebb eltéréseket összevonja (a gráfban ezek kis buborékokként jelennek meg), a harmadik egy haplotípust ad vissza, végül kérhetjük, hogy mindkét haplotípust adja vissza.
Mi nyúl genom összeszerelésre használtuk. A cég szerint elsősorban humán genomra fejlesztették, de annál nagyobb, vagy erősen repetitív genomokra nem javasolják. A mi esetünkben ez nem áll fenn, ezért nyugodtan használhattuk. Ismét csak az a kérdés, hogy a valóság mennyire adja vissza a brosúrák marketing szagú eredményeit, ezért az egyik könyvtárat összeraktam Discovar-denovóval is.
A scaffoldok száma Discovar-denovóval 240 ezer volt, ez lecsökkent 173 ezerre, ha Supernovat használtunk. Az N50 drámai különbségeket mutatott. A bárkódot nem használó módszer csak 1500 körüli hosszúságot adott, míg a másikkal ez az érték 50 ezer lett. Amikor visszaillesztettem a scaffoldokat a genomra, lefedték annak 91%-t!
Viszont észrevettem egy másik érdekes dolgot is. A legtöbb scaffold egymáshoz közel illeszkedett a genomra, kevesebb, mint egy read távolságra. Arra gondoltam, ezeket még közönséges paired-end szekvenálással is össze lehetne rakni.
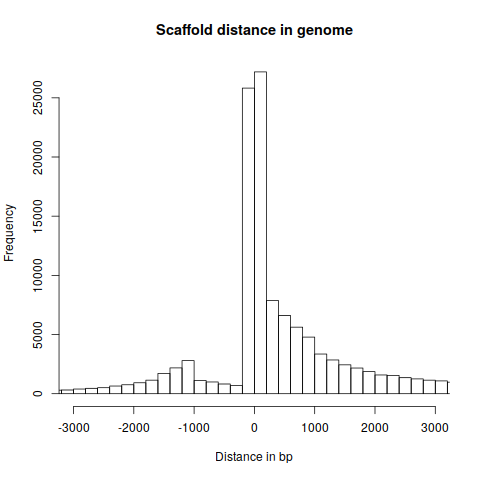
Az egyik hipotézisem az, hogy ezek a nagy fragmentek, amelyek határán már más bárkód van, ezért a program itt megáll. A másik hipotézis, hogy kópiaszám változások vannak ezeken a helyeken, amelyek eltérnek az anyai és apai kromoszómákon.
Ez utóbbinak kicsit ellenmond, hogy a fenti eloszlás akkor is megmarad, ha csak azokra a scaffoldokra rajzolom ki, amelyek méretbeli eltérést mutatnak az anyai és apai kromoszómákon.
Végül a RaGOO-val könnyedén összeraktam őket. A nem-kromoszómális darabok itt sem illeszkedtek a kromoszómákra, viszont kaptunk új régiókat. Ebben az is közrejátszott, hogy másik nyúlfajtát raktunk össze.
A 10x módszere valószínűleg a legjobb megoldás eukarióta draft genomok összerakására. A szoftver nem éri el egy céges termék színvonalát, de az akadémiai bioinformatikai programok közül nem lóg ki. Az eredmény megbízható, de mindenképp szükség van további munkára, A rendszerben még van potenciál, lehet fejleszteni.