
Egyik nap a munkatársaim azon dolgoztak, hogy olyan szkriptet írjanak, ami párhuzamosan elindítja a munkafolyamatokat, figyelik, melyik áll le hibával, stb. Gyakorlatilag egy feladat ütemezőt írtak. Miután rászántak több napot a szkript megírására, még ráment rengeteg idő, hogy a hibákat javítsák, teszteljék.
Ezek azok a programok, amelyeket teljesen felesleges megírni. Miért? Mert már létezik rájuk megoldás. Korábban írtam is róluk, de az idő előre haladtával a dolgok változtak. Lássuk most a legújabb kedvencemet a Snakemake-t.
A Snakemake első ránézésre egy Makefile-szerű rendszer, amivel hierarchikus munkafolyamatokat írhatunk le, de Python nyelven. Ez utóbbi azt jelenti, hogy bárhova beszúrhatunk Python kódot, azt a rendszer végrehajtja. Mivel a bioinformatikában szinte minden lépés fájlok pattogtatását vonja maga után, ezt teszi a Snakemake is. Figyeli, mely kimenetek, mely bemenetekhez tartoznak. Ez határozza meg, hogy mely lépések követik egymást.
Lássunk egy példát, ami a GATK néhány funkcióját valósítja meg. A teljes folyamatból csak azokat a lépéseket szedtem ki, amelyek a Snakemake egy-egy speciális funkciójával kapcsolatosak, és így segítenek megérteni ezt a programot. Egy átlagos GATK futtatás - rendkívül sematikusan - úgy néz ki, hogy először a BAM fájlokat dolgozzuk fel. Beállítjuk a minőségi mutatókat és a fájlokat olyan formára hozzuk, hogy a GATK szeresse. Ezután több BAM fájl eredményét kombináljuk, hogy a variációkat hatékonyabban találjuk meg. Végül ezen a kombinált VCF fájlon végzünk további műveleteket.
A folyamat leírása négy kötelező elemből épül fel: szabály neve, bemeneti fájlok, kimeneti fájlok, feladat.
rule sort:
input:
output:
shell:
Mivel Pythonról van szó (Makefile örökséggel), a sor elején a behúzás kötelező. A rule kulcsszóval nevezzük el a lépést (jelen esetben sort), majd megadjuk a paramétereket. Ha például a BAM fájlokat sorba szeretnénk rendezni, akkor valami ilyesmit kell írnunk:
rule sort:
input:
"raw.bam"
output:
"sorted.bam"
shell:
"samtools sort -o {output} {input}"
Gyakorlatilag ez az alapja a rendszernek. A definíciók közönséges Python sztringek, a lépések között adhatunk Python kódot a rendszerhez. Például tegyük fel, hogy több lépés is igényli a referencia genomot. Létrehozhatunk neki egy változót.
reference = "/mnt/local/nfs/slowmachine/otherexporteddrive/raid/sshmount/nooneknowswhereitis/hg18.fa"
rule genotype:
input:
"vcf/genotype.gvcf.gz"
output:
"vcf/raw.vcf.gz"
shell:
"gatk GenotypeGVCFs -R %s -V {input} -O {output}" % (reference)
De nem muszáj shell szkripteket sem futtatni. A shell kulcsszó helyett, ha run-t használunk, akkor a rendszer a kódot fogja végrehajtani. Például a CombineGVCFs lépés a GATK-ban megköveteli, hogy minden egyes bemeneti fájl előtt legyen egy -V. Ezt nem tudjuk megoldani a Snakemake álltal biztosított hagyományos keretek között.
run:
param = ""
for i in input:
param += " -V " + i
cmd = "gatk CombineGVCFs %s -O {output} -R %s " % (param, reference)
shell(cmd)
Még az sem baj, ha R kódot akarunk futtatni. A run helyett a script kulcsszó segítségével külső Python, R vagy Julia szkripteket futtathatunk.
Természetesen a bemeneti és kimeneti fájlok száma nem korlátozódik egyre. Az ApplyVQSR lépésnél például három bemeneti fájlra van szükségünk.
rule applysnp:
input:
tr="vcf/second12.snp.tr",
recal="vcf/second12.snp.recal",
vcf="vcf/second12.raw.vcf.gz"
output:
"vcf/second12.snpready.vcf.gz"
shell:
"gatk ApplyVQSR -R %s -V {input.vcf} -O {output} --tmp-dir tmp/ --recal-file {input.recal} --tranches-file {input.tr} --mode SNP --truth-sensitivity-filter-level 95.0" % (reference)
Az egyes bemeneti állományokat névvel tudjuk megjelölni. Ugyan ez a módszer használható a kimeneti fájlok esetén.
A bemeneti és kimeneti állományok kezelése hasonló a Makefile-hoz. A Snakemake megvizsgálja, hogy a kineneti fájl létezik-e és újabb-e a bemeneti fájlnál. Ha igen, akkor a lépést nem hajtja végre. Ha nem (mert például módosításokat végeztünk rajta), végrehajtja a lépést. Amennyiben hiba történik egy lépésnél, akkor a csonka kimeneti fájlokat törli. Azért, hogy nyomon követhessük a lépéseket, szükségünk van napló fájlokra. Mivel ezeket nem célszerű törölni egy hibás futás során, azokat a log kulcsszóval definiálhatjuk, az input és output kulcsszavakhoz hasonlóan.
Ezen kívül még több kulcsszó is van. Megadhatjuk például a szükséges erőforrásokat is, hogy a Snakemake hatékonyan tudja futtatni a lépéseket. Erről még lesz szó.
Az eddigi példák csak egy fájlt használtak ki- és bemenetnek. De előfordulhat, hogy ugyan azt a lépést sok fájlon kell végrehajtani. Ilyenkor használjuk a helyettesítő neveket (a doksiban wildcards néven utalnak rá). A helyettesítő nevek hasonlóan működnek, mint a '*' a BASH-ben, de annál érzékenyebb paraméterezést tesznek lehetővé. Visszatérve a sorba rendezős lépésre, ha minden BAM fájlon végre akarjuk hajtani a rendezést, akkor a következő módosításra lesz szükségünk. Tegyük hozzá a naplózást is, valamint jelezzük a használni kívánt szálak számát is.
rule sort:
input: "bam/{sample}.bam"
output: "bam/{sample}.sort.bam"
threads: 8
log: "bam/{sample}.sort.log"
shell:
"samtools sort -@ {threads} -m 4G -o {output} {input} 2>{log}"
Ez alapján a Snakemake még nem tudja, hogyan helyettesítse be a {sample} nevet. Kell egy lépés, ahol ezt meghatározzuk, méghozzá egy olyan lépés során, ahol összefutnak a szálak (vagy elágaznak, attól függően, hogy a folyamat elejét vagy végét nézzük). Ez a mi esetünkben a CombineGVCFs lépés, mert az illesztésekből keletkező GVCF fájlok itt válnak eggyé.
PATIENTS = ["mother", "father", "sick_child", "healthy1", "healthy2"]
rule combine:
input:
expand("{sample}.gvcf.vcf.gz", sample = PATIENTS)
output:
"combined.gvcf.vcf.gz"
A parancs részét már korábban bemutattam, ezért azt nem írnám le még egyszer. A sample helyettesítő név jöhet például a PATIENTS tömbből, de akár az összes fájlt is kiolvashatjuk egy könyvtárból a glob_wildcards parancs segítségével.
Miuán összeállt a munkafolyamat, azt futtatni kell. A snakemake parancs megkeresi a Snakefile fájlt az aktuális könyvtárban, és lefuttatja az abban található első szabályt. Amennyiben a szabály feltétele egy másik szabály, akkor azt is lefuttatja, egészen addig, amig vagy nincs kész a kimeneti fájl, vagy nincs több szabály.
A szabályok hierarchiáját a --dag opcióval kiírhatjuk, ami a mi esetünkben így fest:
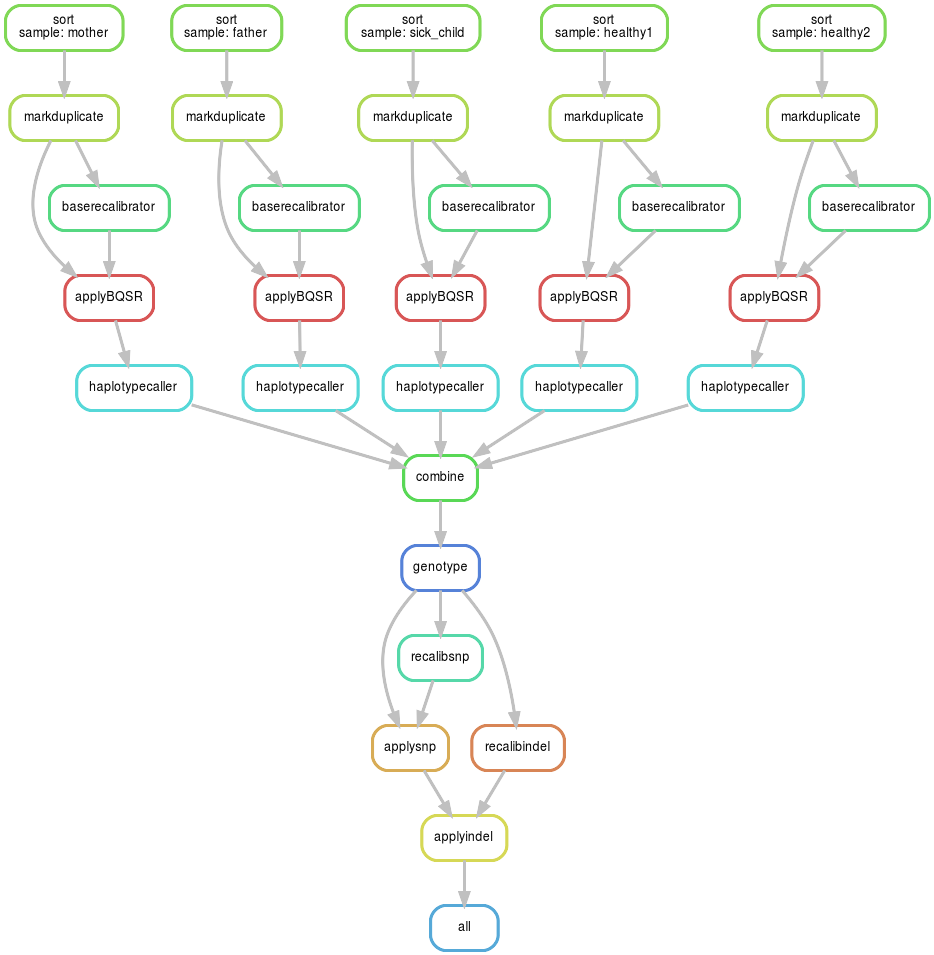
Megjegyezném, hogy ez nem egy valós futás, mert a GATK-nak minimum 10 teljes genom kell, hogy a genotipizálás hatékony legyen, de az nagyon áttekinthetetlen ábrát eredményez, ezért egyszerűsítettem le. A program felismeri, mely lépéseket tudja párhuzamosan végrehajtani, ezért érdemes a szálakat beállítani neki. Képes feladat ütemezőket is használni, sőt különböző felhős infrastruktúrákat, dockert, kubernetet is. (Talán még döglött kutyán is fut.)
Nagyon hasznos funkció, hogy van benne tesztelő üzemmód. A -n kapcsolóval nem hajt végre egyetlen lépést sem, de ellenőrzi a ki- és bemeneti állományokat. Ha hibát talál, jelzi. Egyetlen hátránya, hogy a Python kódot sem futtatja ilyenkor, tehát azt nem tudjuk meg, hogy a kódbetétek szintaxisa megfelelő-e.
Tehát mielőtt elkezdenéd írni a következő Halálcsillag vezérlő szkriptedet, hogy összefogd a rakoncátlan programokat, gondolkozz el rajta: tényleg szükség van rá?